2. Arzneimittel-Zulassung und -Nutzenbewertung in anderen Ländern
Die Zulassung
In Deutschland hängt die Möglichkeit eines Herstellers, ein neues Arzneimittel zu verkaufen und erstattet zu bekommen, zunächst nur von der Zulassung des Mittels ab.
In den anderen Ländern der EU, aber auch anderen Staaten wie etwa den USA, sehen die Anforderungen des Zulassungsprozesses in weiten Bereichen sehr ähnlich aus: Ein neues Arzneimittel wird auf die Parameter pharmazeutische Qualität, Sicherheit und Wirksamkeit und Verträglichkeit überprüft. In kontrollierten Studien muss das Mittel mindestens besser sein als ein Placebo. Abweichungen von diesem Schema gibt es zum Beispiel bei beschleunigten Zulassungsverfahren, die nur unter besonderen Voraussetzungen beschritten werden können.
Ein wichtiger Schritt für eine über die EU hinausgehende internationale Harmonisierung der Zulassungsverfahren war die Einführung eines gemeinsamen Dossiers (Common Technical Document, CTD), um eine Zulassung zu beantragen. Dieses wird seit 2003 in Europa, in Japan und in den USA verbindlich eingesetzt.
Trotz dieser Harmonisierung ist es möglich, dass nationale Behörden zu unterschiedlichen Ergebnissen kommen. Ein Mittel kann beispielsweise in den USA zugelassen, in der EU hingegen nicht zugelassen werden. Dies hängt u. a. von Interpretationsspielräumen ab, ob ein Mittel pharmazeutisch gut, sicher, wirksam und verträglich ist.
Die Nutzenbewertung
Darüber hinaus haben Länder in den letzten drei Jahrzehnten aber auch weitere Parameter und Prozesse für die Entscheidung über einen Marktzugang und/oder eine Kostenübernahme entwickelt, die über die drei Hürden Qualität, Sicherheit und Wirksamkeit/Verträglichkeit hinausgehen.
Dahinter steht zum einen die Beobachtung, dass Kostenträger mit immer stärker steigenden Gesundheitskosten konfrontiert, die Mittel für Ausgaben indes begrenzt sind. Zum anderen behaupten Hersteller zwar in der Regel, dass neue Mittel besser sind als die vorhandenen und damit zu Recht teurer; diese Behauptungen lassen sich jedoch oft nicht bestätigen, wie Untersuchungen wiederholt belegen (Stichwort: Scheininnovation). Die meisten Studien sehen den Anteil wirklicher Innovationen sogar bei unter 15 Prozent.
Gesundheitssysteme haben darauf mit zwei – teils ineinander verwobenen – Ansätzen reagiert. In der einen Gruppe von Ländern beruht die Entscheidung für den Marktzugang und die Kostenübernahme weiterhin auf rein medizinischen Nutzenparametern.
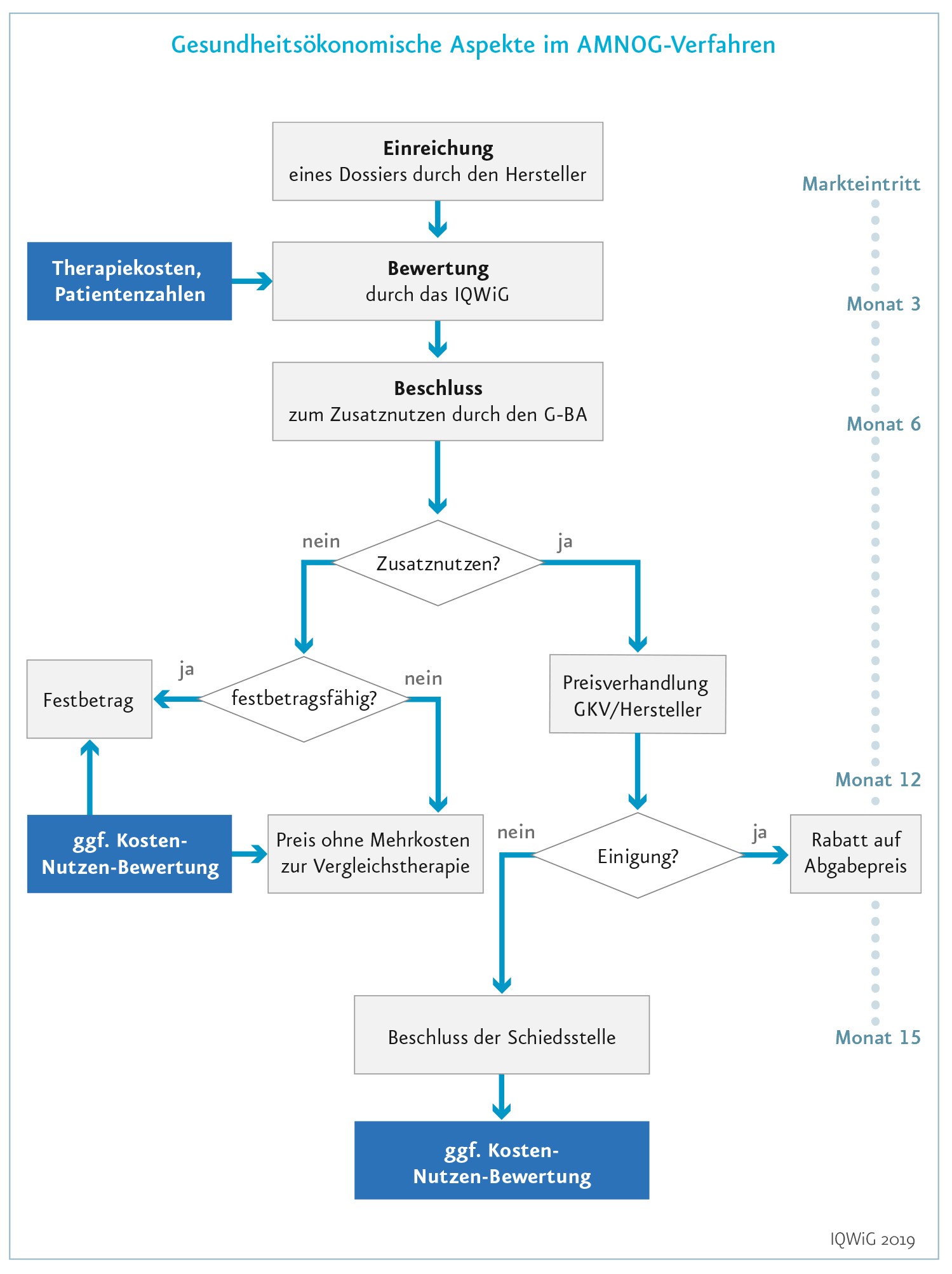
Zusätzlich zum klassischen Zulassungsverfahren durchlaufen neue Arzneimittel weitere Verfahren (wie in Deutschland die bereits beschriebene frühe Nutzenbewertung), in denen ein Mittel seinen postulierten Mehrwert im Vergleich zu einer Standardbehandlung beweisen muss und nicht nur im Vergleich zu einem Placebo. Die Daten aus diesen Vergleichsstudien müssen einen Zusatznutzen in Endpunkten belegen, die für Patienten wichtig sind, wie etwa eine gesenkte Sterblichkeit ( Mortalität), eine verminderte Krankheitslast ( Morbidität) oder eine erhöhte Lebensqualität. Vom Hersteller geforderte höhere Kosten werden nur dann übernommen, wenn es tatsächlich einen Zusatznutzen gibt.
Länder mit diesem Ansatz sind zum Beispiel Frankreich, Österreich, Dänemark, Italien, Spanien, Schweiz, Türkei oder Südkorea.
Kosteneffektivität
In einer zweiten Gruppe von Ländern spielen nicht nur rein medizinische Parameter eine Rolle, sondern es wird auch auf der Grundlage gesundheitsökonomischer Analysen über eine Kostenübernahme entschieden.
Den Anfang machte 1993 Australien, gefolgt von Kanada 1994. Weitere Länder, in denen eine Kosteneffektivitätsanalyse in den Entscheidungsprozess einbezogen wird, sind England, Wales, Schottland und Neuseeland.
Manche Länder arbeiten dabei mit einem Schwellenwert, also einem Verhältnis von Kosten pro zusätzlicher Nutzeneinheit, das nicht überschritten werden darf. Als Nutzeneinheit im Nenner dieses Verhältnisses wird häufig das sogenannte qualitätskorrigierte Lebensjahr oder QALY (engl. quality-adjusted life year) eingesetzt. In dieser Kennzahl werden qualitative Bewertungen von Gesundheitszuständen mit quantitativen Angaben zur Dauer dieser Zustände kombiniert. Eine Verlängerung des Überlebens oder eine Verbesserung der gesundheitsbezogenen Lebensqualität erhöht den Wert; was das Leben verkürzt oder die Lebensqualität beeinträchtigt, verringert ihn. Ein Vorteil des QALY: Das Maß ist generisch, macht also den Nutzen von Therapien über verschiedene Indikationen hinweg vergleichbar.
Dieses Verfahren wird indes nicht überall gleichermaßen stringent durchgeführt. In Australien kann ein Überschreiten des Schwellenwerts dazu führen, dass ein Mittel nicht in die Positivliste aufgenommen wird, also die Liste der Arzneimittel, deren Kosten vom staatlichen Gesundheitssystem übernommen werden. In den Niederlanden kommt es auch bei einem noch so ungünstigen Kosten-Nutzen-Verhältnis fast nie zu einer Ablehnung einer Erstattung. Und in England gibt es nach einer negativ ausgegangenen Kosteneffektivitätsanalyse alternative Erstattungsmöglichkeiten, auf die zurückgegriffen werden kann, häufig nach einer ausgeprägten Diskussion über die Kostenübernahme in der Öffentlichkeit.
In Deutschland gibt es in der Regel keine Kosteneffektivitätsanalyse für neue Arzneimittel. Sollten sich Krankenkassen und Hersteller im Verlauf der Preisverhandlungen jedoch nicht auf einen Erstattungsbetrag einigen, können auch gesundheitsökonomische Parameter ins Spiel kommen, um doch noch eine Einigung zu erzielen. Schwellenwerte kommen dabei aber nicht zum Einsatz.

Die vierte Hürde
Wird der Marktzugang oder die Erstattungsfähigkeit eines Medikaments von zusätzlichen Kriterien neben Qualität, Sicherheit und Wirksamkeit/Verträglichkeit abhängig gemacht, spricht man von einer "vierten Hürde". Diese besteht in einigen Ländern wie Frankreich im Nachweis eines Zusatznutzens, in anderen wie Australien in einem Nachweis der Kosteneffektivität.
Die frühe Nutzenbewertung in Deutschland ist keine vierte Hürde, da Medikamente ohne belegten Zusatznutzen am Markt bleiben können. Die Kosten des Arzneimittels sollen dann aber nicht höher sein als die der Standardbehandlung.

Spezialfall USA
In den USA wird der Listenpreis für ein neues Medikament nach der Zulassung durch die amerikanische Arzneimittelbehörde FDA allein durch die Hersteller festgesetzt, was in Deutschland nur für das erste Jahr möglich ist. Es gibt in den USA keine staatliche Instanz und keine andere Institution, die diesen Preis für das gesamte Land aushandeln oder festlegen könnte. Stattdessen handeln zahlreiche private Krankenversicherer, Apotheken- oder Klinikketten und Großhändler – teils über sogenannte Pharmacy Benefit Manager in externen Firmen – direkt mit den Herstellern Rabatte für neue Arzneimittel aus. Die Hersteller können zudem die Preise ihrer Arzneimittel jährlich oder sogar halbjährlich erhöhen. Dies führte zum Beispiel dazu, dass von 2012 bis 2016 der Preis für mehr als drei Viertel der bestverkauften Arzneimittel um mehr als 50 Prozent anstieg, teils ohne ersichtlichen klinischen Grund (siehe Robinson 2020).
Auch die staatlichen Krankenversorgungssysteme wie Medicare (für ältere und behinderte Menschen) und Medicaid (für ärmere Personen) bzw. die Krankenversicherung der Veteranen der US-Armee handeln mit den Herstellern Rabatte aus. Während bei Medicare Kosten überhaupt keine Rolle spielen, fordern die Vertreter der Veteranenorganisation eine Kosteneffektivitätsanalyse, die auf QALYs beruhen kann, aber nicht muss.
Dieses System trägt dazu bei, dass in den USA die Kosten für Arzneimittel so hoch sind wie in keinem vergleichbaren Staat, etwa den OECD-Ländern. Befürworter wenden ein, dass es nur aufgrund der hohen Arzneimittelpreise möglich sei, so viele innovative Arzneimittel zu entwickeln wie in den USA.
Neue Arzneimittel: Zulassung, Nutzenbewertung, Erstattung
1. Arzneimittel-Zulassung und frühe Nutzenbewertung in Deutschland
2. Arzneimittel-Zulassung und -Nutzenbewertung in anderen Ländern
3. Das AMNOG-Verfahren: mehr als nur Kostenkontrolle
4. Besondere Therapierichtungen und traditionelle Arzneimittel


{kind=link}